Overview of my PhD projects
 Notre Dame
Notre Dame
Multiphoton microscopy (MPM)
Multiphoton microscopy (MPM) is a widely used in vivo imaging technique in biological and medical applications. In the case of two-photon excitation fluorescence (2PEF) microscopy, two photons excite a fluorophore, which in turn emits a single higher-energy photon. The rate of 2PEF depends quadratically on the excitation intensity, thus enabling the axial localization of excitation in the vicinity of the focal plane. The 2PEF generally uses near-infrared (NIR) excitation, which experiences less scattering in tissue than shorter-wavelength light and does not cause background autofluorescence. A comparison between single-photon and 2PEF can be seen in the figure below, where 2PEF has a much higher localization capability. Therefore, out-of-focus fluorescence is avoided, and the overall photobleaching and phototoxicity in thick samples are reduced.

Fluorescence lifetime imaging microscopy (FLIM)
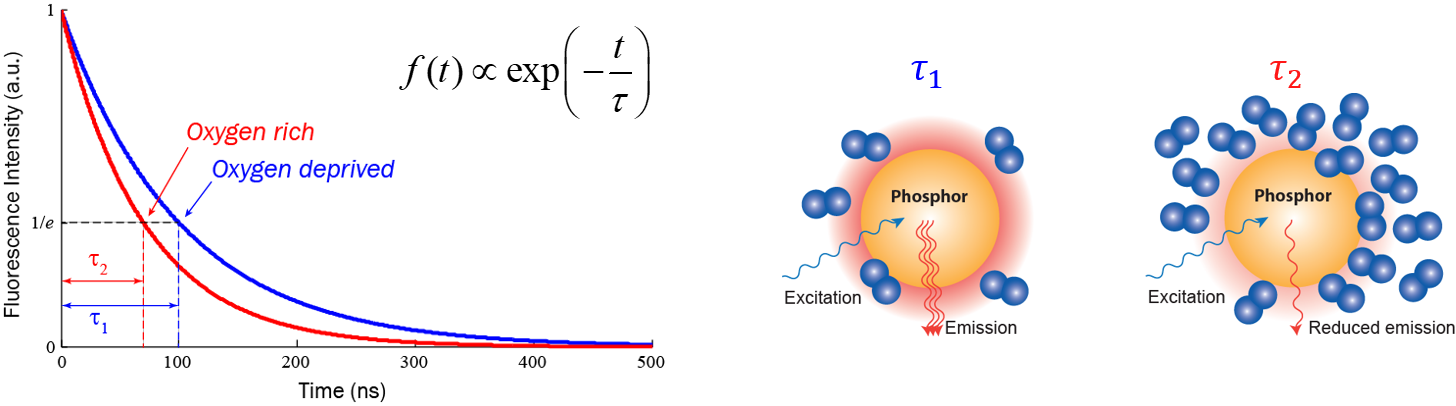
Fluorescence lifetime imaging microscopy (FLIM) is also a powerful tool in biological, chemical, and medical studies: it provides an additional contrast, the fluorescence decay lifetime of excited fluorophores, in fluorescence microscopy. FLIM can be employed to image local calcium or potassium ion concentrations, dissolved oxygen concentrations, pH, refractive index, or the occurrence of fluorescence resonance energy transfer. For example, the figure below shows how fluorescence lifetime changes following dissolved oxygen concentration in the microenvironment: the lifetime in an oxygen rich environment is obviously shorter than the lifetime in an oxygen deprived environment; hence by measuring fluorescence lifetime, one can extract dissolved oxygen concentration in the microenvironment after a calibration. Compared with conventional intensity-based microscopy techniques, FLIM has the advantage of significantly reduced sensitivity to errors caused by non-uniform optical
absorption, scattering, photobleaching, and fluctuations in fluorophore concentration.
Super-sensitivity MPM-FLIM
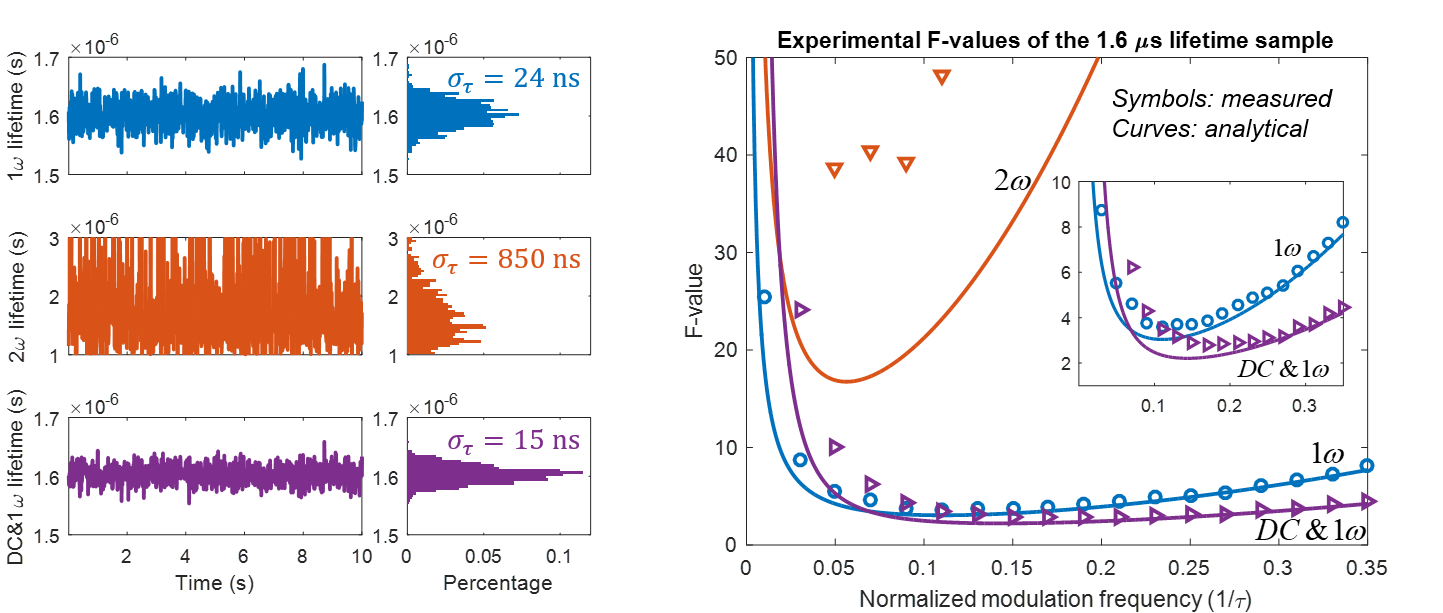
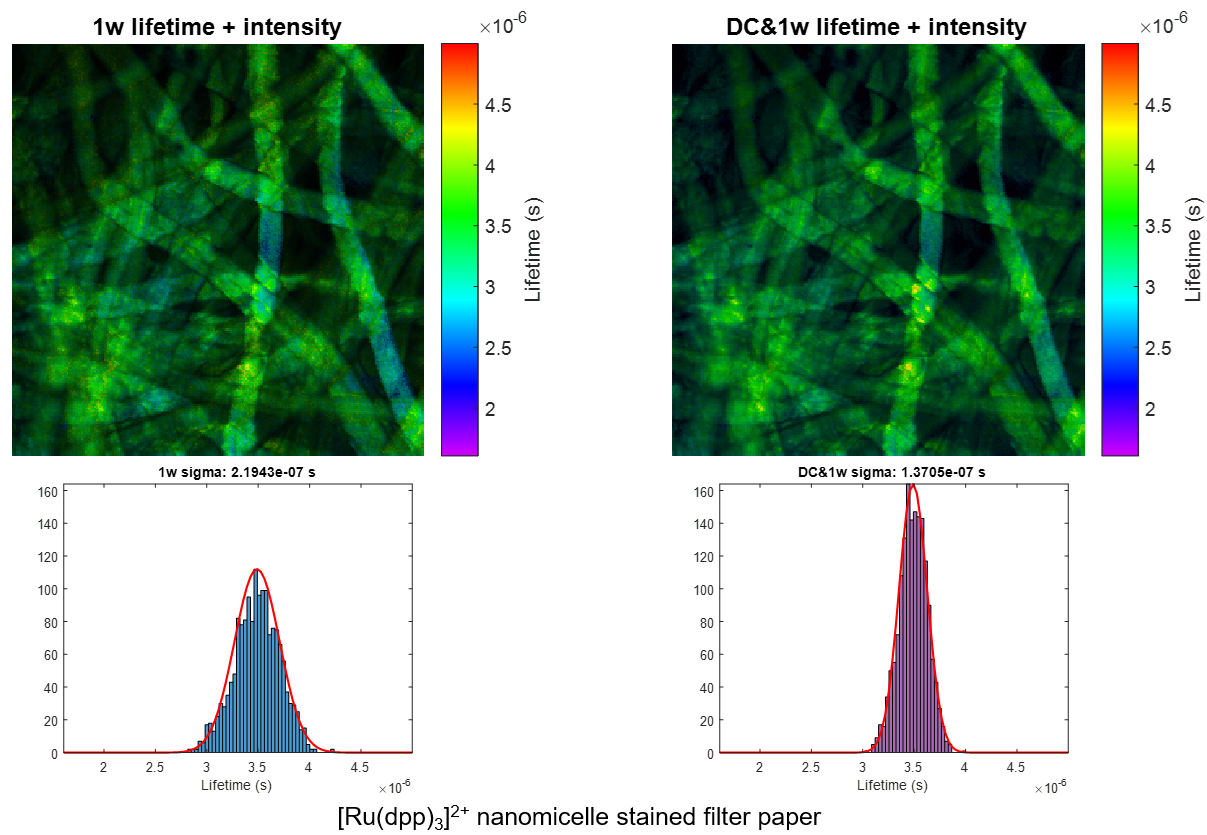
We present a series of experiments that demonstrate a super-sensitive chemical imaging technique based on multiphoton frequency-domain fluorescence lifetime imaging microscopy (MPM-FD-FLIM) that shows a 2x improvement in imaging speed compared to the theoretical limit of conventional MPM-FD-FLIM. Additionally, this technique produces unprecedented sensitivity over a large range of fluorescence lifetimes. These results are achieved through simple modifications to data analysis in a conventional MPM-FD-FLIM microscope and are based on an analytical model describing the signal-to-noise ratio (SNR) of an MPM-FD-FLIM system [J. Opt. Soc. Am. A 33, B1 (2016)].
Related publications
- Yide Zhang, Aamir A. Khan, Genevieve D. Vigil, and Scott S. Howard, “Investigation of signal-to-noise ratio in frequency-domain multiphoton fluorescence lifetime imaging microscopy”, Journal of the Optical Society of America A, vol. 33, no. 7, pp. B1-B11, July 2016, DOI: 10.1364/JOSAA.33.0000B1.
- Yide Zhang, Aamir A. Khan, Genevieve D. Vigil, and Scott S. Howard, “Super-sensitivity multiphoton frequency-domain fluorescence lifetime imaging microscopy”, Optics Express, vol. 24, no. 18, pp. 20862-20867, Sept. 2016, DOI: 10.1364/OE.24.020862.
- Yide Zhang, Genevieve D. Vigil, Aamir A. Khan, and Scott S. Howard, “Doubling the sensitivity of multiphoton frequency-domain fluorescence lifetime images”, CLEO: Science and Innovations 2017, San Jose, California USA, May 2017, DOI: 10.1364/CLEO_SI.2017.SM3C.6.
Saturation-compensated FLIM
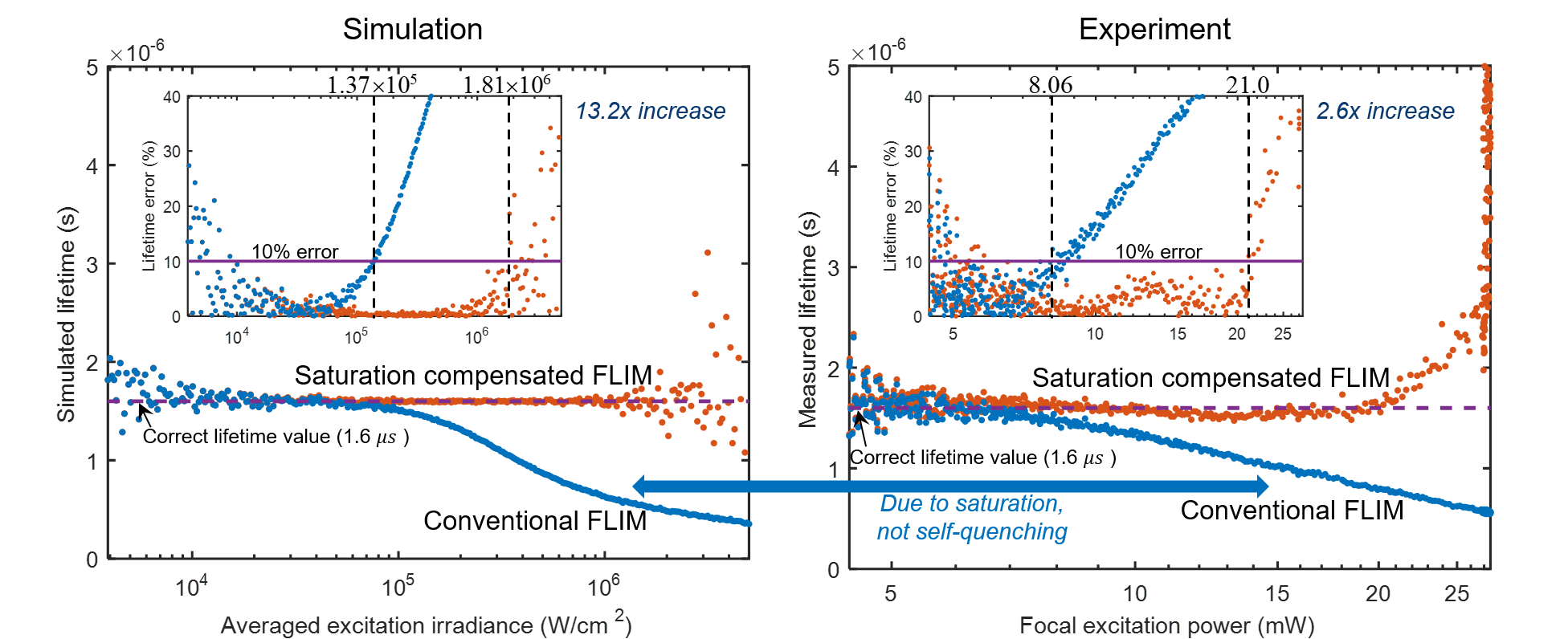
Fluorophore saturation is the key factor limiting the speed and excitation range of fluorescence lifetime imaging microscopy (FLIM). For example, fluorophore saturation causes incorrect lifetime measurements when using conventional frequency-domain FLIM at high excitation powers. We present an analytical theoretical description of this error and present a method for compensating for this error in order to extract correct lifetime measurements in the limit of fluorophore saturation. We perform a series of simulations and experiments to validate our methods. The simulations and experiments show a 13.2× and a 2.6 × increase in excitation range, respectively. The presented method is based on algorithms that can be easily applied to existing FLIM setups.

Related publications
- Yide Zhang, Genevieve D. Vigil, Lina Cao, Aamir A. Khan, David Benirschke, Tahsin Ahmed, Patrick Fay, and Scott S. Howard, “Saturation-compensated measurements for fluorescence lifetime imaging microscopy”, Optics Letters, vol. 42, no. 1, pp. 155-158, Jan. 2017, DOI: 10.1364/OL.42.000155.
Super-resolution microscopy: stepwise optical saturation (SOS)
Super-resolution fluorescence microscopy is an important tool in biomedical research for its ability to discern features smaller than the diffraction limit. However, due to its difficult implementation and high cost, the super-resolution microscopy is not feasible in many applications. In this paper, we propose and demonstrate a saturation-based super-resolution fluorescence microscopy technique that can be easily implemented and requires neither additional hardware nor complex post-processing. The method is based on the principle of stepwise optical saturation (SOS), where M steps of raw fluorescence images are linearly combined to generate an image with a $\sqrt{M}$-fold increase in resolution compared with conventional diffraction-limited images. For example, linearly combining (scaling and subtracting) two images obtained at regular powers extends the resolution by a factor of 1.4 beyond the diffraction limit. The resolution improvement in SOS microscopy is theoretically infinite but practically is limited by the signal-to-noise ratio. We perform simulations and experimentally demonstrate super-resolution microscopy with both one-photon (confocal) and multiphoton excitation fluorescence. We show that with the multiphoton modality, the SOS microscopy can provide super-resolution imaging deep in scattering samples.
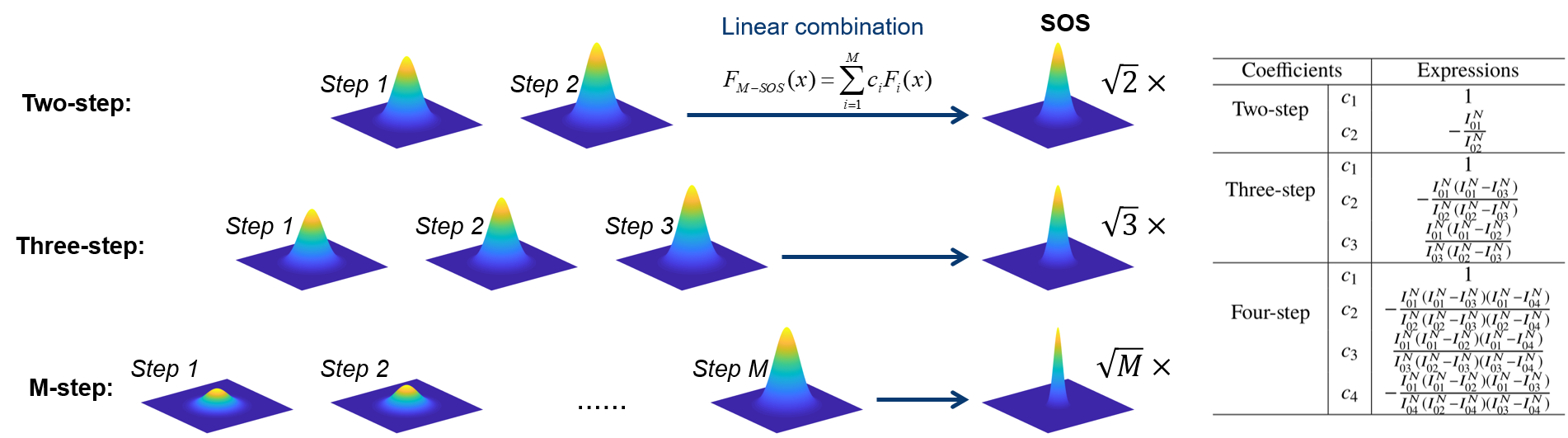
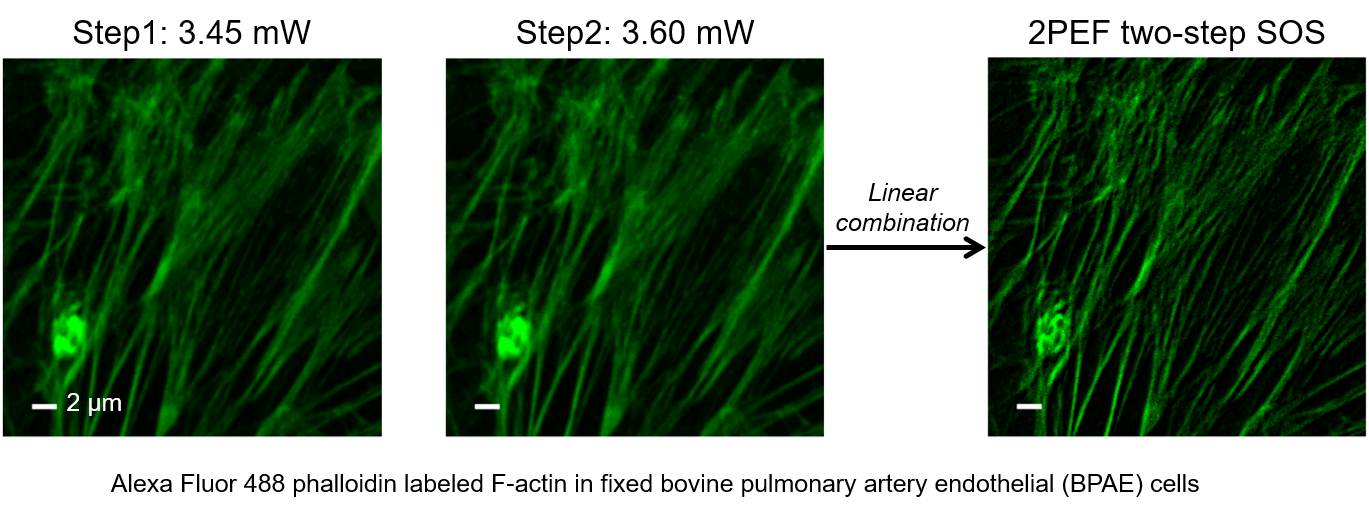
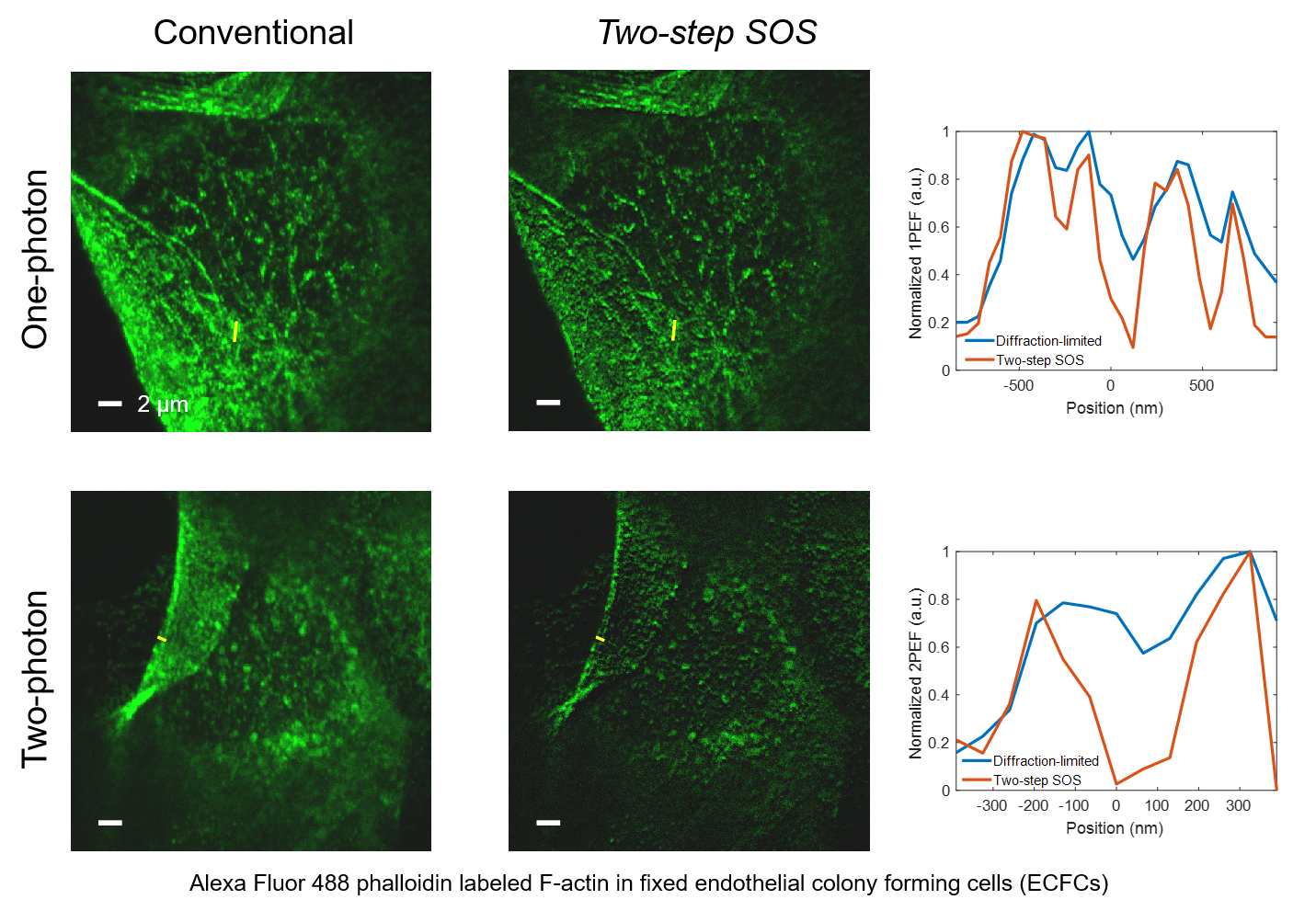
Related publications
- Yide Zhang, Prakash D. Nallathamby, Genevieve D. Vigil, Aamir A. Khan, Devon E. Mason, Joel D. Boerckel, Ryan K. Roeder, and Scott S. Howard, “Super-resolution fluorescence microscopy by stepwise optical saturation”, Biomedical Optics Express, vol. 9, no. 4, pp. 1613-1629, Apr. 2018, DOI: 10.1364/BOE.9.001613.
- Yide Zhang, David Benirschke, and Scott S. Howard, “Stepwise optical saturation microscopy: obtaining super-resolution images with conventional fluorescence microscopes”, Biomedical Optics 2018, Hollywood, Florida USA, Apr. 2018, DOI: 0.1364/TRANSLATIONAL.2018.JTh3A.27.
Super-resolution FLIM: generalized stepwise optical saturation (GSOS)
We present a novel super-resolution fluorescence lifetime microscopy technique called generalized stepwise optical saturation (GSOS) that generalizes and extends the concept of the recently demonstrated stepwise optical saturation (SOS) super-resolution microscopy [Biomed. Opt. Express 9, 1613 (2018)]. The theoretical basis of GSOS is developed based on exploring the dynamics of a two-level fluorophore model and using perturbation theory. We show that although both SOS and GSOS utilize the linear combination of M raw images to increase the imaging resolution by a factor of √ M, SOS is a special and the simplest case of GSOS. The super-resolution capability is demonstrated with theoretical analysis and numerical simulations for GSOS with sinusoidal and pulse-train modulations. Using GSOS with pulse-train modulation, super-resolution and fluorescence lifetime imaging microscopy (FLIM) images can be obtained simultaneously. The super-resolution FLIM capability is experimentally demonstrated with a cell sample on a custom-built two-photon frequency-domain (FD) FLIM system based on radio frequency analog signal processing. To our knowledge, this is the first implementation of super-resolution imaging in FD-FLIM.
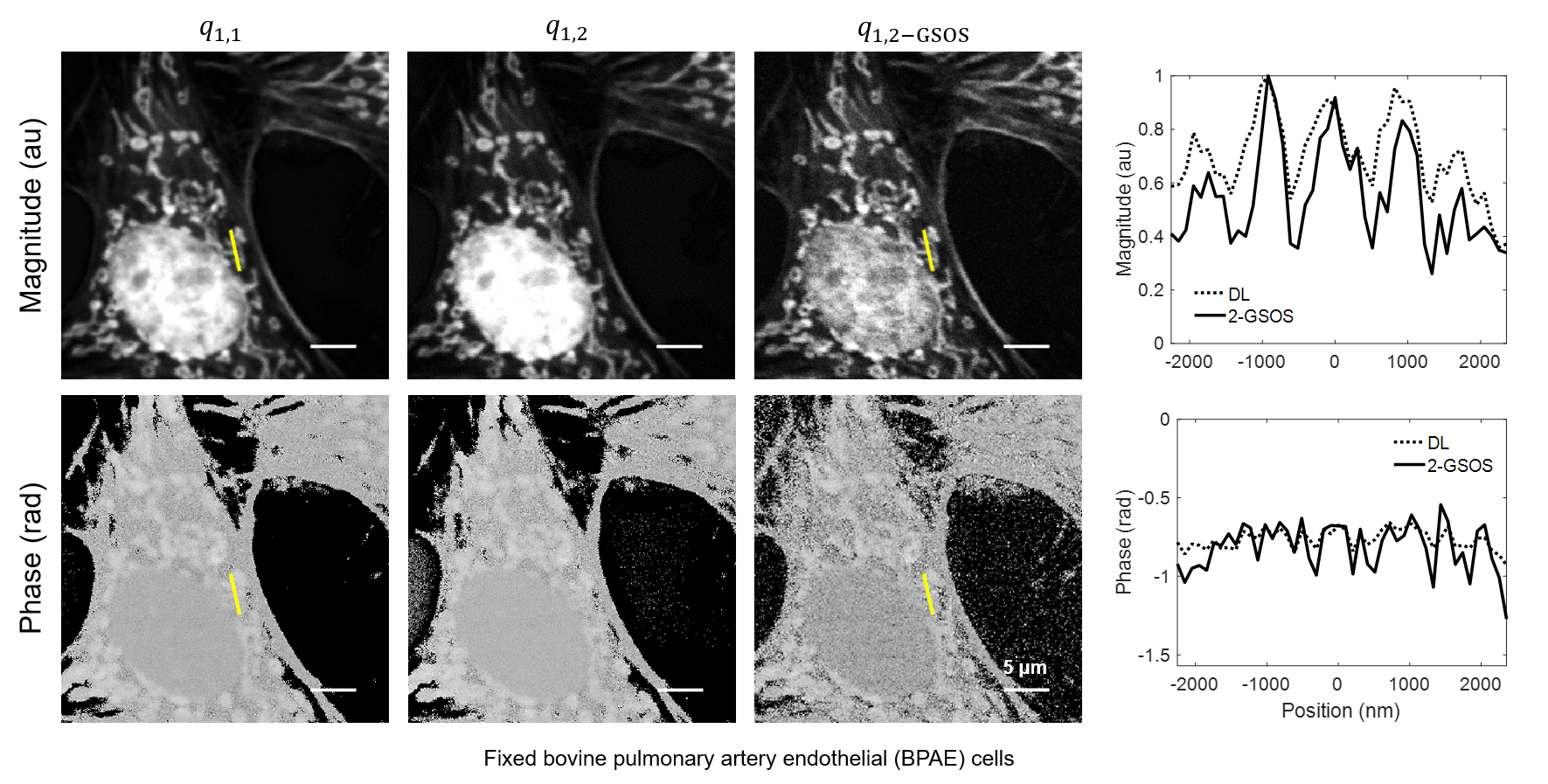
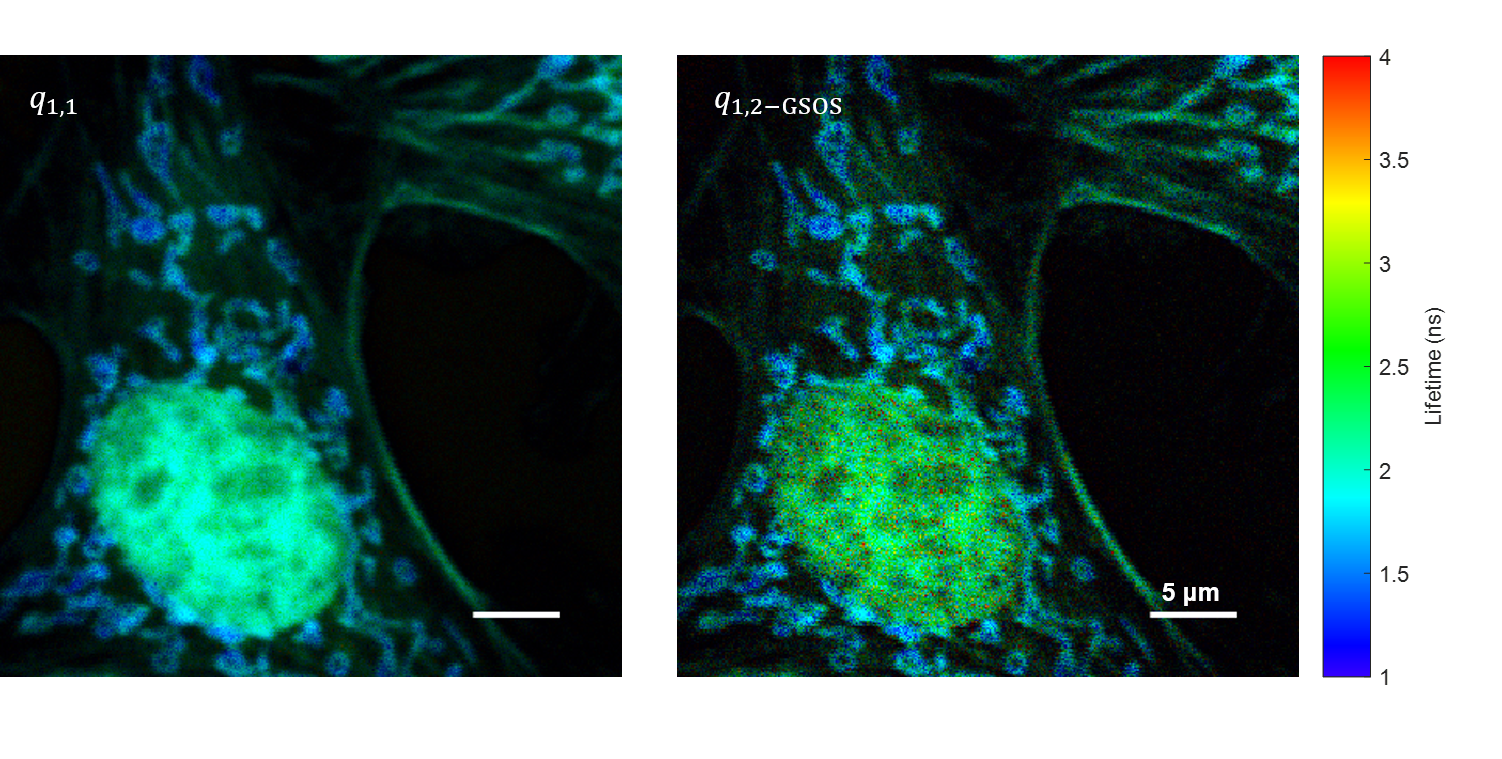
Related publications
- Yide Zhang, David Benirschke, Ola Abdalsalam, and Scott S. Howard, “Generalized stepwise optical saturation enables super-resolution fluorescence lifetime imaging microscopy”, Biomedical Optics Express, vol. 9, no. 9, pp. 4077-4093, Sept. 2018, DOI: 10.1364/BOE.9.004077.
- Yide Zhang, David Benirschke, Ola Abdalsalam, and Scott S. Howard, “Super-resolution multiphoton frequency-domain fluorescence lifetime imaging microscopy by generalized stepwise optical saturation (GSOS)”, SPIE Photonics West 2019, San Francisco, California USA, Feb. 2019, DOI: 10.1117/12.2507663.
Super-resolution microscopy: deconvolution stepwise optical saturation (DeSOS)
Super-resolution microscopy is broadening our in-depth understanding of cellular structure. However, super-resolution approaches are limited, for numerous reasons, from utilization in longer-term intravital imaging. We devised a combinatorial imaging technique that combines deconvolution with stepwise optical saturation microscopy (DeSOS) to circumvent this issue and image cells in their native physiological environment. Other than a traditional confocal or two-photon microscope, this approach requires no additional hardware. Here, we provide an open-access application to obtain DeSOS images from conventional microscope images obtained at low excitation powers. We show that DeSOS can be used in time-lapse imaging to generate super-resolution movies in zebrafish. DeSOS was also validated in live mice. These movies uncover that actin structures dynamically remodel to produce a single pioneer axon in a “top-down” scaffolding event. Further, we identify an F-actin population – stable base clusters – that orchestrate that scaffolding event. We then identify that activation of Rac1 in pioneer axons destabilizes stable base clusters and disrupts pioneer axon formation. The ease of acquisition and processing with this approach provides a universal technique for biologists to answer questions in living animals.
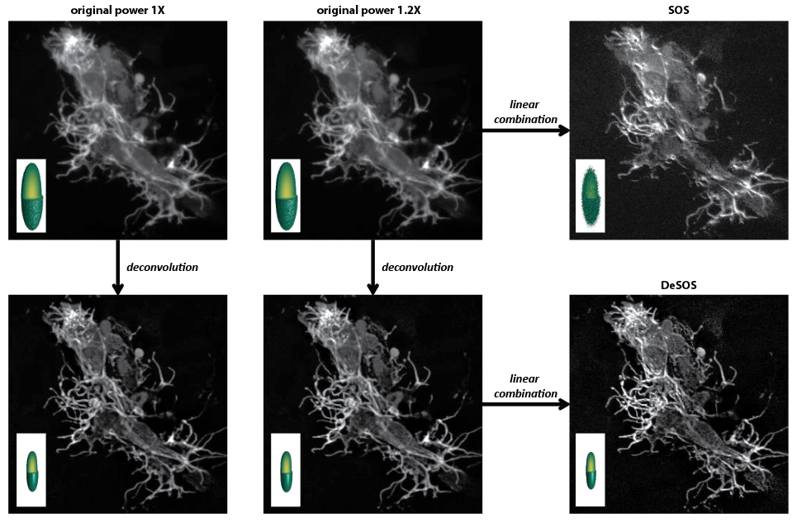
Related publications
- Yide Zhang, Evan L. Nichols, Abigail M. Zellmer, Ian H. Guldner, Cody Kankel, Siyuan Zhang, Scott S. Howard, and Cody J. Smith, “Generating intravital super-resolution movies with conventional microscopy reveals actin dynamics that construct pioneer axons”, Development, vol. 146, no. 5, pp. dev171512, Feb. 2019, DOI: 10.1242/dev.171512.
Fluorescence Microscopy Denoising (FMD) dataset and deep learning denoising methods
Fluorescence microscopy has enabled a dramatic development in modern biology. Due to its inherently weak signal, fluorescence microscopy is not only much noisier than photography, but also presented with Poisson-Gaussian noise where Poisson noise, or shot noise, is the dominating noise source, instead of Gaussian noise that dominates in photography. To get clean fluorescence microscopy images, it is highly desirable to have effective denoising algorithms and datasets that are specifically designed to denoise fluorescence microscopy images. While such algorithms exist, there are no such datasets available. In this paper, we fill this gap by constructing a dataset - the Fluorescence Microscopy Denoising (FMD) dataset - that is dedicated to Poisson-Gaussian denoising. The dataset consists of 12,000 real fluorescence microscopy images obtained with commercial confocal, two-photon, and wide-field microscopes and representative biological samples such as cells, zebrafish, and mouse brain tissues. We use imaging averaging to effectively obtain ground truth images and 60,000 noisy images with different noise levels. We use this dataset to benchmark 10 representative denoising algorithms and find that deep learning methods have the best performance. To our knowledge, this is the first microscopy image dataset for Poisson-Gaussian denoising purposes and it could be an important tool for high-quality, real-time denoising applications in biomedical research.
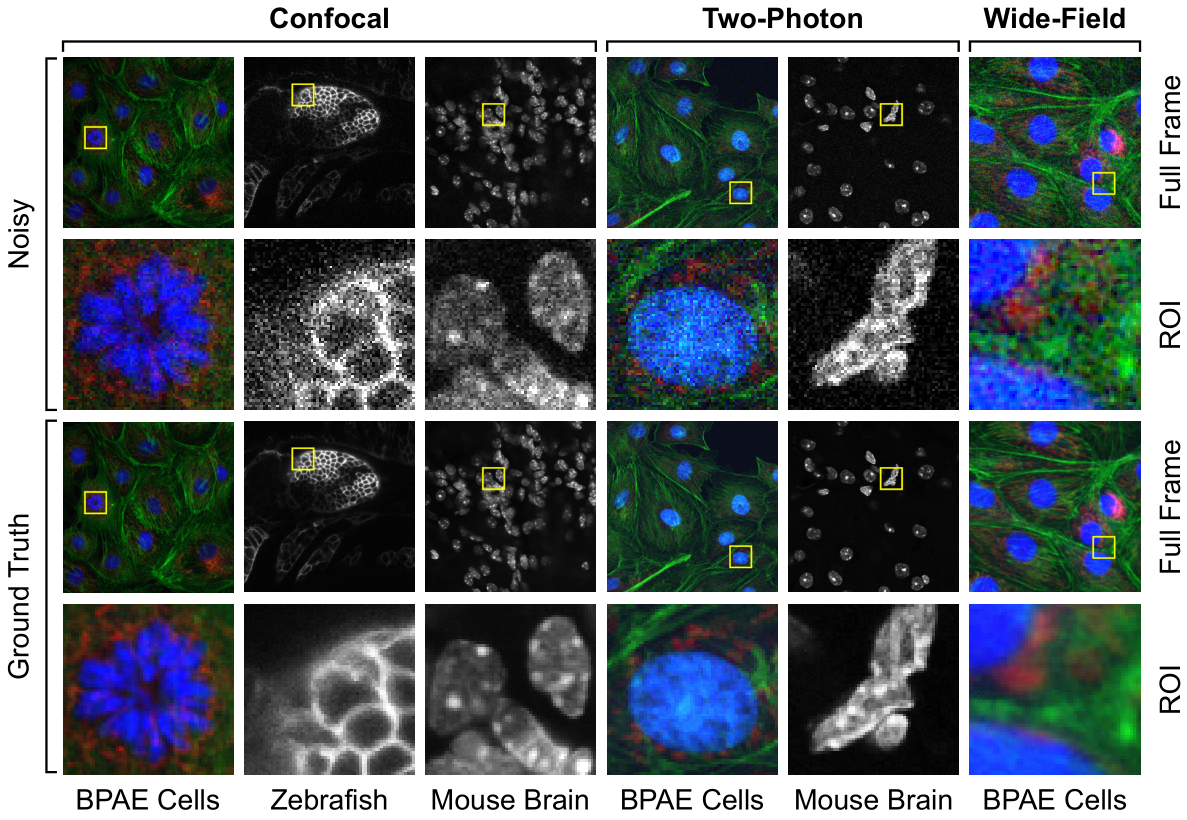
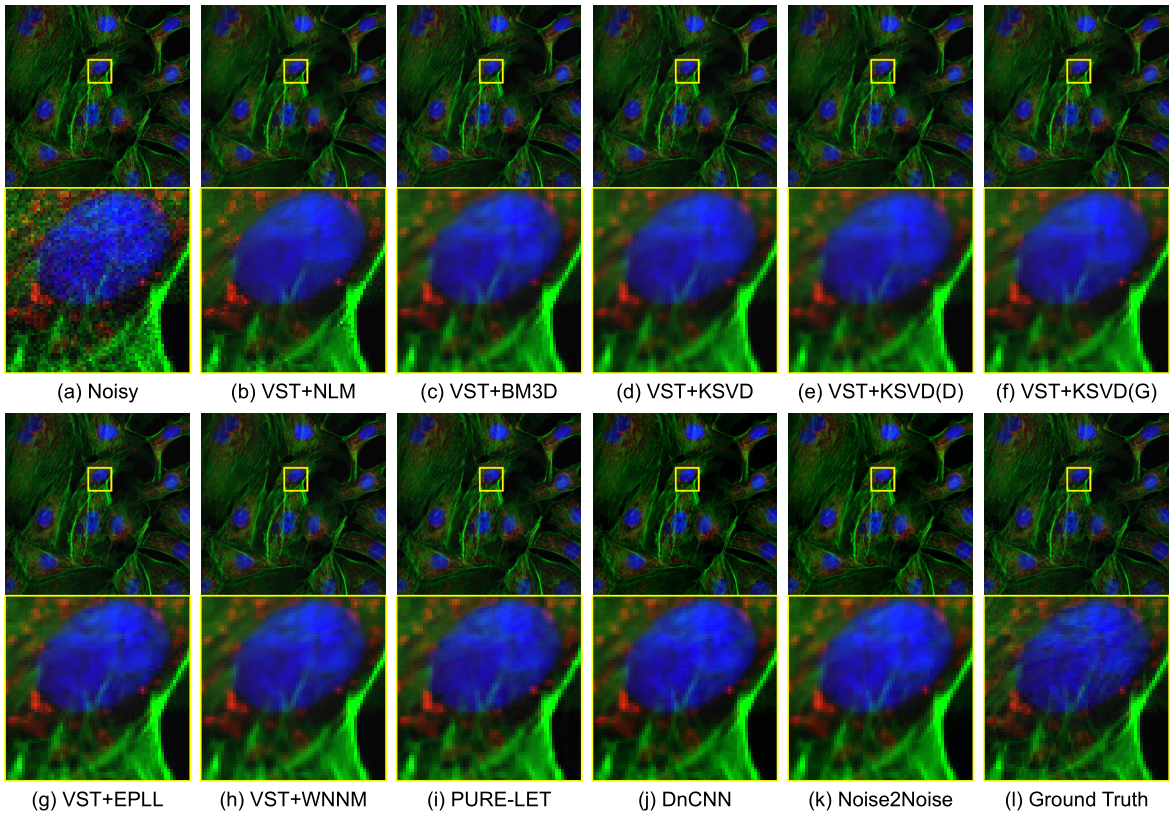
Related publications
- Yide Zhang, Yinhao Zhu, Evan Nichols, Qingfei Wang, Siyuan Zhang, Cody Smith, and Scott Howard, “A Poisson-Gaussian denoising dataset with real fluorescence microscopy images”, IEEE/CVF Conference on Computer Vision and Pattern Recognition (CVPR 2019), Long Beach, California USA, June 2019, DOI: 10.1109/CVPR.2019.01198.
Phase-multiplexing FLIM and phasor plots
We propose and demonstrate a novel multiphoton frequency-domain fluorescence lifetime imaging microscopy (MPM-FD-FLIM) system that is able to generate 3D lifetime images in deep scattering tissues. The imaging speed of FD-FLIM is improved using phase multiplexing, where the fluorescence signal is split and mixed with the reference signal from the laser in a multiplexing manner. The system allows for easy generation of phasor plots, which not only address multi-exponential decay problems but also clearly represent the dynamics of the fluorophores being investigated. Lastly, a sensorless adaptive optics setup is used for FLIM imaging in deep scattering tissues. The capability of the system is demonstrated in fixed and living animal models, including mice and zebrafish.
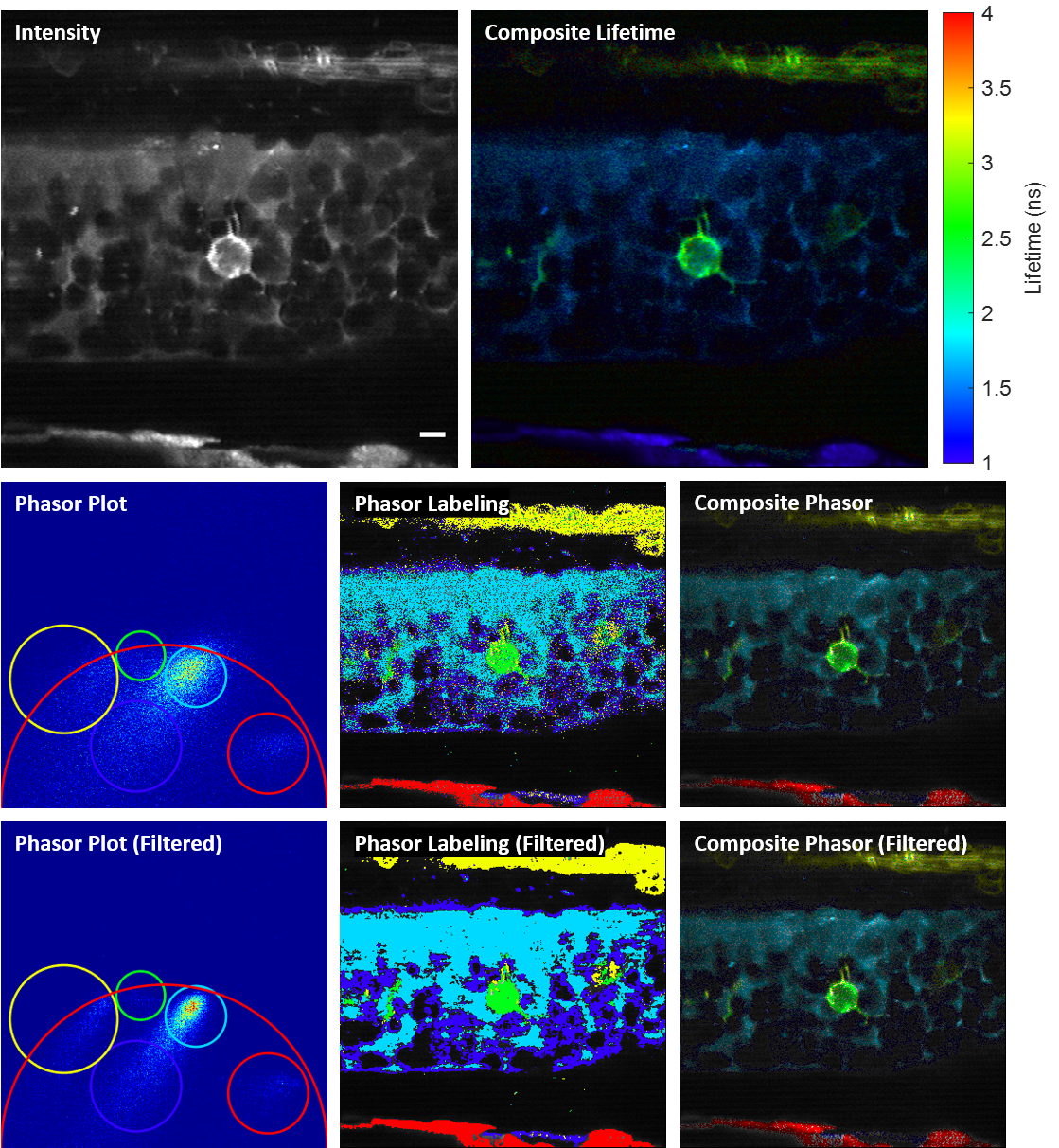
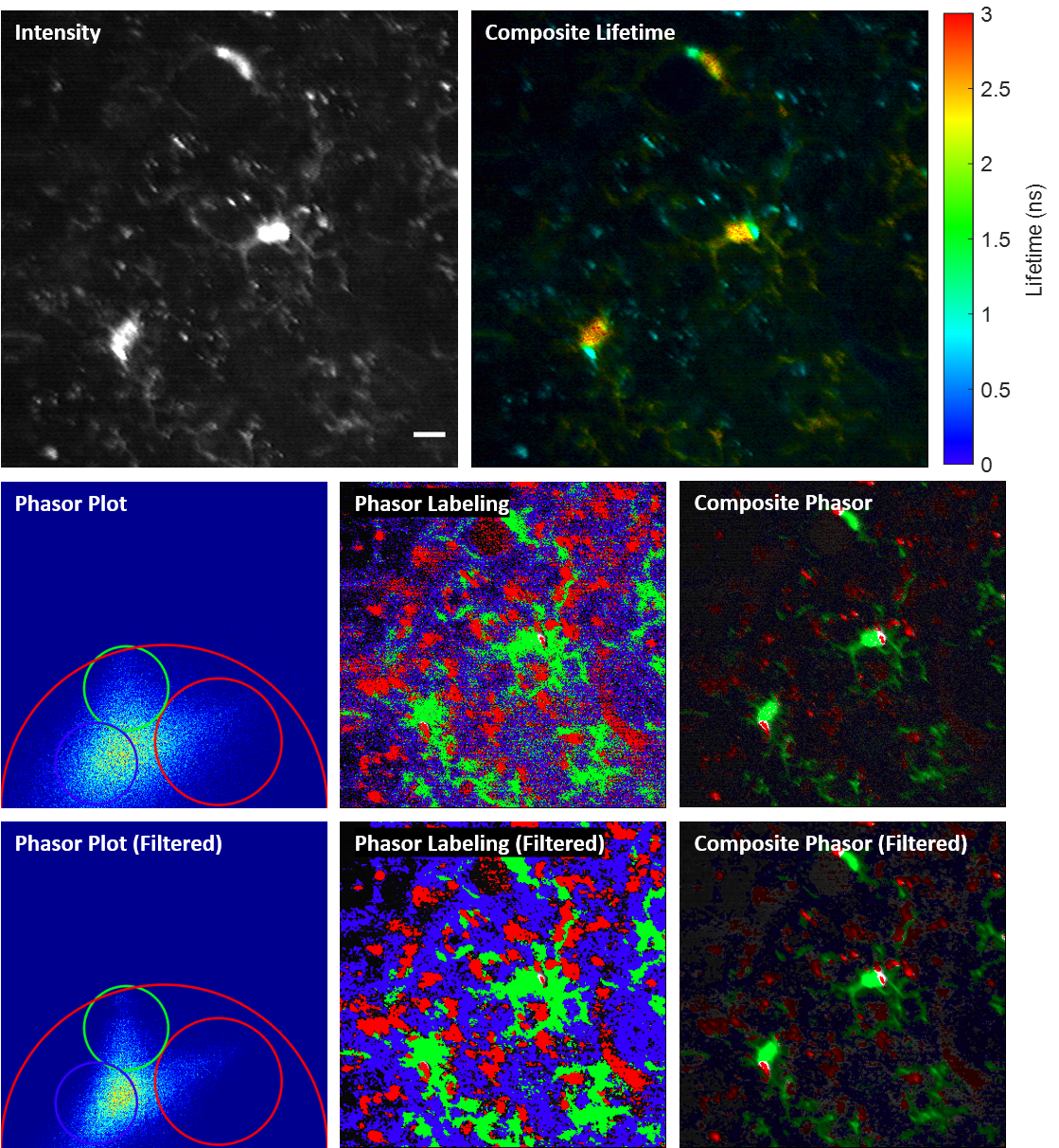
Related publications
- Yide Zhang, Ian H. Guldner, Evan L. Nichols, David Benirschke, Cody J. Smith, Siyuan Zhang, and Scott S. Howard, “Three-dimensional deep tissue multiphoton frequency-domain fluorescence lifetime imaging microscopy via phase multiplexing and adaptive optics”, SPIE Photonics West 2019, San Francisco, California USA, Feb. 2019, DOI: 10.1117/12.2510674.
Automatic segmentation of FLIM images
Fluorescence lifetime imaging microscopy (FLIM) provides additional contrast for fluorophores with overlapping emission spectra. The phasor approach to FLIM greatly reduces the complexity of FLIM analysis and enables a useful image segmentation technique by selecting adjacent phasor points and labeling their corresponding pixels with different colors. This phasor labeling process, however, is empirical and could lead to biased results. In this Letter, we present a novel and unbiased approach to automate the phasor labeling process using an unsupervised machine learning technique, i.e., K-means clustering. In addition, we provide an open-source, user-friendly program that enables users to easily employ the proposed approach. We demonstrate successful image segmentation on 2D and 3D FLIM images of fixed cells and living animals acquired with two different FLIM systems. Finally, we evaluate how different parameters affect the segmentation result and provide a guideline for users to achieve optimal performance.
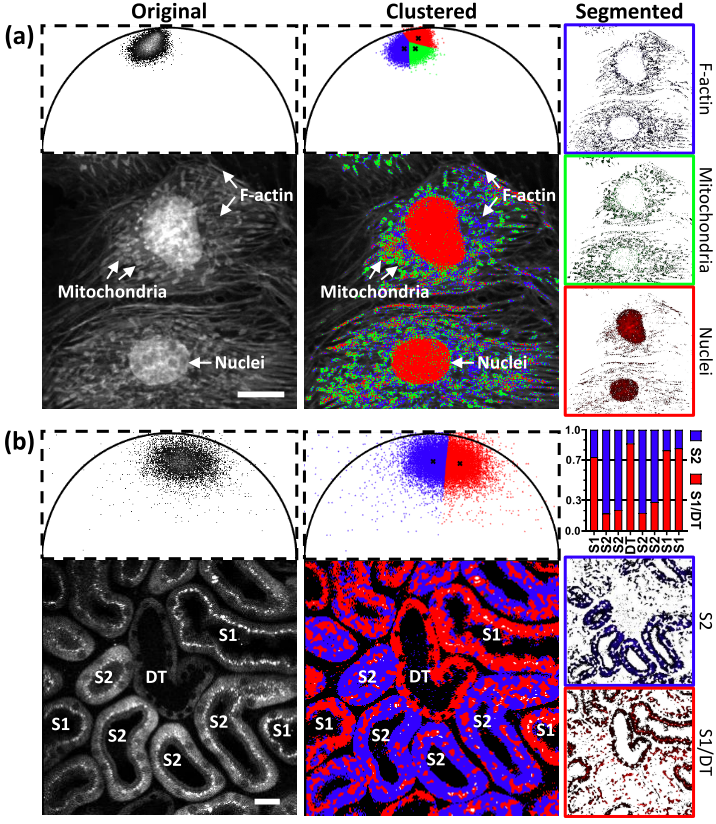
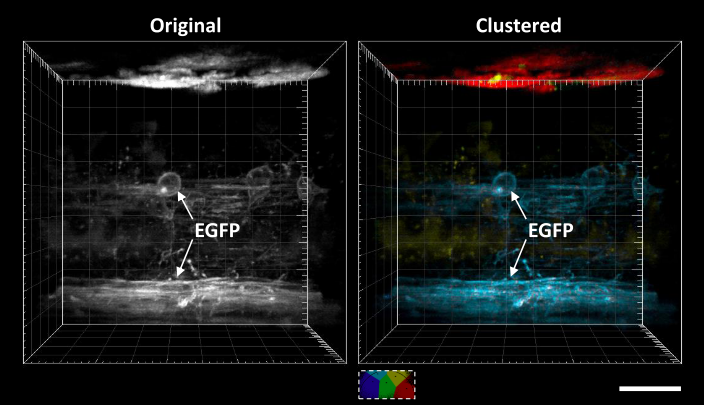
Related publications
- Yide Zhang, Takashi Hato, Pierre C. Dagher, Evan L. Nichols, Cody J. Smith, Kenneth W. Dunn, and Scott S. Howard, “Automatic segmentation of intravital fluorescence microscopy images by K-means clustering of FLIM phasors”, Optics Letters, vol. 44, no. 16, pp. 3928-3931, Aug. 2019, DOI: 10.1364/OL.44.003928.
Resolution enhancement of transmission electron microscopy (TEM) images
Super-resolution fluorescence microscopy techniques have enabled dramatic development in modern biology due to their capability to discern features smaller than the diffraction limit of light. Recently, super-resolution radial fluctuations (SRRF), an analytical approach that is capable of generating super-resolution images easily without the need for specialized hardware or photoswitchable fluorophores, has been presented. While SRRF has only been demonstrated on fluorescence microscopes, in principle, this method can be used to generate super-resolution images on any imaging platforms with intrinsic radial symmetric point spread functions. In this work, we show that SRRF can be utilized to enhance the resolution and quality of transmission electron microscopy (TEM) images. By including an image registration algorithm to correct for sample drift, the SRRF-TEM approach substantially enhances the resolution of TEM images of three different samples acquired with a commercial TEM system. We quantify the resolution improvement in SRRF-TEM and evaluate how SRRF parameters affect the resolution and quality of SRRF-TEM results.
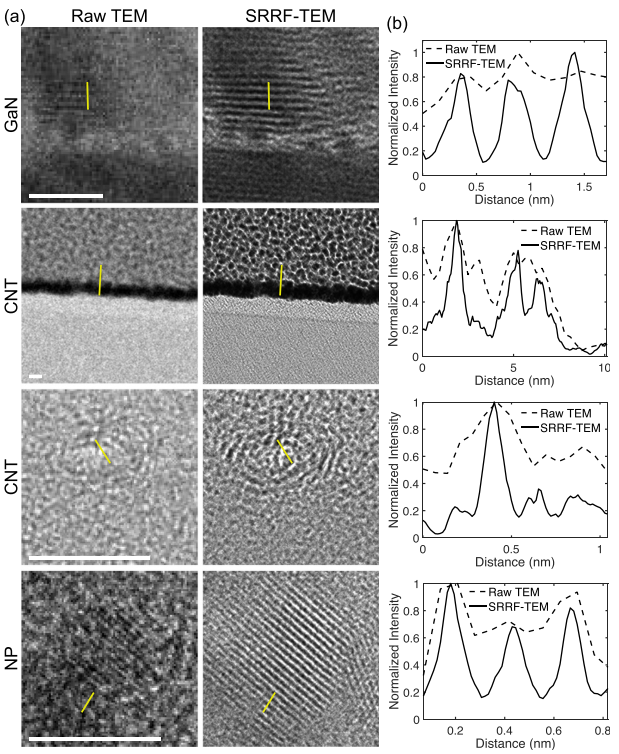
Related publications
- Yide Zhang, Sergei Rouvimov, Xiaotong Yuan, Karla Gonzalez-Serrano, Alan C. Seabaugh, and Scott S. Howard, “Resolution enhancement of transmission electron microscopy by super-resolution radial fluctuations”, Applied Physics Letters, vol. 116, no. 4, pp. 044105, Jan. 2020, DOI: 10.1063/1.5128353.
Yide Zhang
NIH K99 Postdoctoral Fellow
My research is interdisciplinary and focused on developing new types of optical imaging techniques that could advance the work of other researchers and medical personnel in a wide variety of fields. Currently, I am developing next-generation photoacoustic and ultrafast imaging techniques that can observe biological and physical phenomena that are too fast to be imaged with existing methods. The observation of the ultrafast phenomena could provide a better understanding of the fundamentals of life and physical sciences. I am also developing novel quantum imaging approaches that can investigate biological organisms with an imaging performance that cannot be achieved using classical optical imaging. In my free time, I enjoy cooking, hiking, cycling, and traveling.